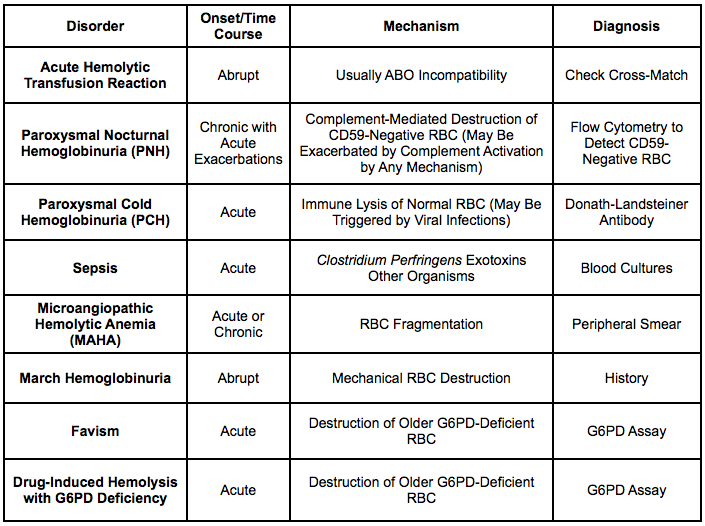
Physiology: deficiency of the glycolipid, glycosylphosphatidyl-inositol (GPI), which anchors CD55 and CD59 to membrane -> increased complement-mediated RBC (and granulocyte/platelet) lysis
Physiologic Mechanisms Which Contribute to Hemolysis in Liver Disease
Acquired Alterations of Red Blood Cell Membrane
Burr Cells (Echinocytes)
Spur Cells (Acanthocytes)
Stomatocytes: mouth-shaped area of central pallor
Target Cells: due to decreased lecithin/cholesterol acyltransferase (LCAT) activity, resulting in increased cholesterol:phospholipid ratio -> absolute increase in surface area of the red blood cell membrane
Hypersplenism (see Splenomegaly): due to portal hypertension
Acquired Acanthocytosis
Acquired Stomatocytosis
Spur Cell Hemolysis
Physiology
Background-Iron Physiology
Iron: free iron is toxic to cells and is therefore stored in alternate forms
Within Cells: iron is complexed to protein, as ferritin or hemosiderin (apoferritin binds to free ferrous iron and stores it in its ferric state)
As ferritin accumulates with cells of the reticuloendothelial system, protein aggregates are formed as hemosiderin -> hemosiderin is less readily available for utilization than ferritin is
In the Circulation: serum iron is bound to transferrin
Background-Red Blood Cell Physiology
Normal Red Blood Cell Lifespan: 110-120 days
Definition of Hemolysis: shortening of RBC survival to <100 days
Normal Rate of Red Blood Cell Age-Independent Random Hemolysis: rate is <0.05-0.5% per day
Hemolysis (Anemia) Results in a Compensatory increase in the Erythropoietin Secretion: increases reticulocyte percentage (and absolute reticulocyte count) -> increases RBC production
Reticulocytosis is Found in Most Patients with Hemolytic Anemia (and Acute Hemorrhage)
Predominant Site of Hemolysis
Intravascular Hemolysis (RBC Destruction Within the Vascular Space)
General Comments
Intravascular Hemolysis Results in Release of Hemoglobin into the Plasma Either as Red Oxyhemoglobin or Brownish Oxidized Methemoglobin: plasma has a red-brown color
Free hemoglobin binds to haptoglobin -> hemoglobin-haptoglobin complex is rapidly removed by the liver (results in decreased serum haptoglobin level)
Lysed hemoglobin breaks down into alpha-beta dimers, which are small (MW: 34k) and cleared via glomerular filtration by the kidney -> resulting in hemoglobinuria
Hemoglobin dimers filtered by the kidney are taken up by renal tubular cells, degraded, and the iron stored as hemosiderin -> when renal tubular cells are later sloughed into the urine (approximately 7 days later), hemosiderinuria can be detected (by the Prussian blue reaction)
Autoimmune Hemolytic Anemia (AIHA): may occur in some cases (where IgM forms an antibody-antigen complex on the RBC membrane -> activation of complement with RBC lysis)
Extravascular Hemolysis (Red Blood Cell Destruction in the Spleen, Other Reticuloendothelial Tissues, or Extravasated Blood/Hematoma)
General Comments
Hepatic Destruction of RBC’s: primary site of destruction of severely damaged RBC’s (especially those coated with complement)
Liver receives larger percentage of cardiac output than the spleen
Splenic Destruction of RBC’s: primary site of destruction of poorly-deformable RBC’s (spherocytes, etc), due to narrow passage in the cords of Billroth -> macrophage ingestion of RBC’s (with degradation of hemoglobin to iron/biliverdin/carbon monoxide)
Biliverdin is converted to unconjugated bilirubin, which is released into the plasma
Autoimmune Hemolytic Anemia (AIHA): usually extravascular (most cases are IgG-mediated)
Physiology: the kidneys do not filter unconjugated bilirubin (as it is avidly bound to albumin) -> therefore, the presence of bilirubinuria indicates the presence of conjugated bilirubinemia
Etiology of Bilirubinuria*
Urobilinogen
Etiology of Increased Urobilinogen: in urine and stool
Physiology: with intravascular hemolysis, hemoglobin is released from hemolyzed RBC’s into the blood, exceeding the binding capacity of haptoglobin -> excess hemoglobin is filtered by the kidney
Some of this hemoglobin is excreted in the urine, resulting in hemoglobinuria (with “coca cola-colored” urine)
Some of this hemoglobin is reabsorbed in the proximal convoluted tubule, where the iron portion is removed and stored in ferritin or hemosiderin -> proximal tubule cells slough off (containing the hemosiderin) and are excreted into the urine, resulting in hemosiderinuria
Urine hemosiderin (composed of a complex of ferritin, denatured ferritin, and other material) can be detected in iron-stained urinary sediment (within the sloughed proximal tubular cells)
Diagnostic Utility
Urine hemoglobin disappears more quickly from the urine than hemosiderin, making it less sensitive for the presence of hemolysis (especially in cases with intermittent hemolysis)
Urine Hemosiderin can remain in the urine for several weeks (making it a more sensitive marker for hemolysis in the recent past): however, after an acute episode of intravascular hemolysis, several days may pass before urinary hemosiderin can be detected
Reticulocytes are Newly-Released RBC’s: they are slightly larger than mature RBC’s and have some residual ribosomal RNA -> presence of RNA allows for staining, with detection and counting
Normal Reticulocyte Percentage: 1-2%
Reticulocyte Production Index (RPI) = Reticulocyte Percentage x (Patient’s Hct/Normal Hct) x (1/RMT): corrects reticulocyte percentage for the degree of anemia (normalized to Hct 45%) and reticulocyte maturation time (RMT)
Use Normal Hct = 45%
RMT
Hct 45% -> RMT = 1.0 days
Hct 15% -> RMT = 2.5 days
Reticulocyte Production Index > or = to 2.5%: indicates adequate bone marrow response to anemia [MEDLINE]
Acute/Subacute Hemorrhage: note that acute hemorrhage may not result in an increased RPI, due to the time that it takes to increase epo synthesis and increase bone marrow RBC production
Hemolytic Anemia
Reticulocyte Production Index <2.5%: indicates inadequate bone marrow response to anemia [MEDLINE]
Chronic Anemia
Hypoproliferative Anemia: such as iron deficiency, marrow hyporesponsiveness, aplasia, etc
Maturation Disorder: such as vitamin B2 deficiency, etc
Definition: smear with precursor cells of the myeloid and erythroid lineage, which usually indicates the presence of extramedullary hematopoiesis (predominantly in the spleen)
Target Cells (Bell-Shaped Codocytes, Mexican Hat Cells)
Definition
Etiology
Asplenia (see Asplenia): splenic macrophages normally clear opsonized, deformed, and damaged red blood cells -> if splenic macrophage function is abnormal/absent because due to splenectomy, altered red blood cells will not be removed from the circulation appropriately
Auto-Splenectomy Resulting from Sickle Cell Disease (see Sickle Cell Disease)
Post-Splenectomy
End-Stage Liver Disease (ESLD) (see End-Stage Liver Disease): decreased lecithin/cholesterol acyltransferase (LCAT) activity, resulting in increased cholesterol:phospholipid ratio -> absolute increase in surface area of the red blood cell membrane
Hemoglobin C Disease
Iron Deficiency Anemia (see Iron Deficiency Anemia): decrease in hemoglobin content relative to red blood cell surface area
Thalassemias (see Thalassemias): decreased hemoglobin content relative to red blood cell surface area
Definition: presence of spherocytes indicates loss of RBC membrane surface area in excess of loss of cell volume -> spherocytes are a feature of many hemolytic anemias
Etiology
Interaction of Membrane-Antibody Complex with Reticuloendothelial System
Skull/Skeletal Deformities: can occur in childhood due to increased hematopoiesis and resultant bone marrow expansion in disorders such as thalassemia
Pulmonary Manifestations
Pulmonary Hypertension (see Pulmonary Hypertension): seen with specific chronic hemolytic anemias
References
General
Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood 1987;69(3):820 [MEDLINE]
Use of trimethoprim-sulfamethoxazole in a glucose-6-phosphate dehydrogenase-deficient population. Rev Infect Dis 1987; 9(Suppl 2):S218-S229 [MEDLINE]
A meta-analysis of salvage therapy for Pneumocystis carinii pneumonia. Arch Intern Med 2001; 25;161(12):1529-1533 [MEDLINE]
Autoimmune Hemolytic Anemia. Am J Hematol. 2002 Apr;69(4):258-71 [MEDLINE]
Second-line salvage treatment of AIDS-associated Pneumocystis jirovecii pneumonia: a case series and systematic review. J Acquir Immune Defic Syndr 2008; 48:63-67 [MEDLINE]
Clindamycin-primaquine versus pentamidine for the second-line treatment of Pneumocystis pneumonia. J Infect Chemother 2009; 15(5):343-346 [MEDLINE]
Clinical efficacy of first- and second-line treatments for HIV- associated Pneumocystis jirovecii pneumonia. A tri-centre cohort study. J Antimicrob Chemother 2009; 64(6):1282-1290 [MEDLINE]
Drug-induced immune haemolytic anaemia in the Berlin Case-Control Surveillance Study. Br J Haematol 2011; 154:644-653. Doi: 10.1111/j.1365-2141.2011.08784.x; First published online 12 July 2011 [MEDLINE]
Etiology
Investigation of the pathogenesis of massive hemolysis in a case of Clostridium perfringens septicemia. Ann Hematol. 1993;67(3):145 [MEDLINE]
Effects of Clostridium perfringens recombinant and crude phospholipase C and theta-toxin on rabbit hemodynamic parameters. J Infect Dis. 1995;172(5):1317 [MEDLINE]